제일원리계산은 물질의 상태를 기술하는 Schrödinger 방정식의 해를 구하는 것이다.
실험없이도 플랑크 상수나 전자의 질량과 같은 기본 상수로부터 물질의 특성을 계산한다.
수소(H)와 같이 단순한 물질은 해석적으로도 풀 수 있지만 분자나 고체와 같은 복잡한 문제는 수치적 접근 외에는 방법이 없다.
따라서 수많은 과학자들은 Schrödinger 방정식을 풀어내기 위해 Born-Oppenheimer(BO) 근사, Hatree-Fock(HF) 근사, Khon-Sham(KS) 방정식을 얻어냈고 지금도 더 나은 근사법들이 개발되고 있다. 그리고 현대에 들어 컴퓨터의 발달로 Schrödinger 방정식의 근에 매우 근접한 수치적인 해를 구할 수 있게 된다.
특히, 제일원리계산은 전자를 직접적으로 기술하기 때문에 컴퓨터가 해낸 이러한 계산은 실험값을 매우 높은 정확도로 예측 했으며, 미지의 물질을 연구하는 것도 가능하게 했다. 그리고 고속연산장치(HPC)가 지속적으로 발전하면서 신소재공학에서 다루는 결함(defect)을 포함한 복잡한 물질을 계산하는 방향으로 확대되고 있다.
또한 머신러닝, 딥러닝과 같은 기법의 발전과 제일원리계산의 융합으로 미지의 물질을 창조하고 신소재에서 필요로 하는 인자 선별이 더욱 가속화 될 전망이다.


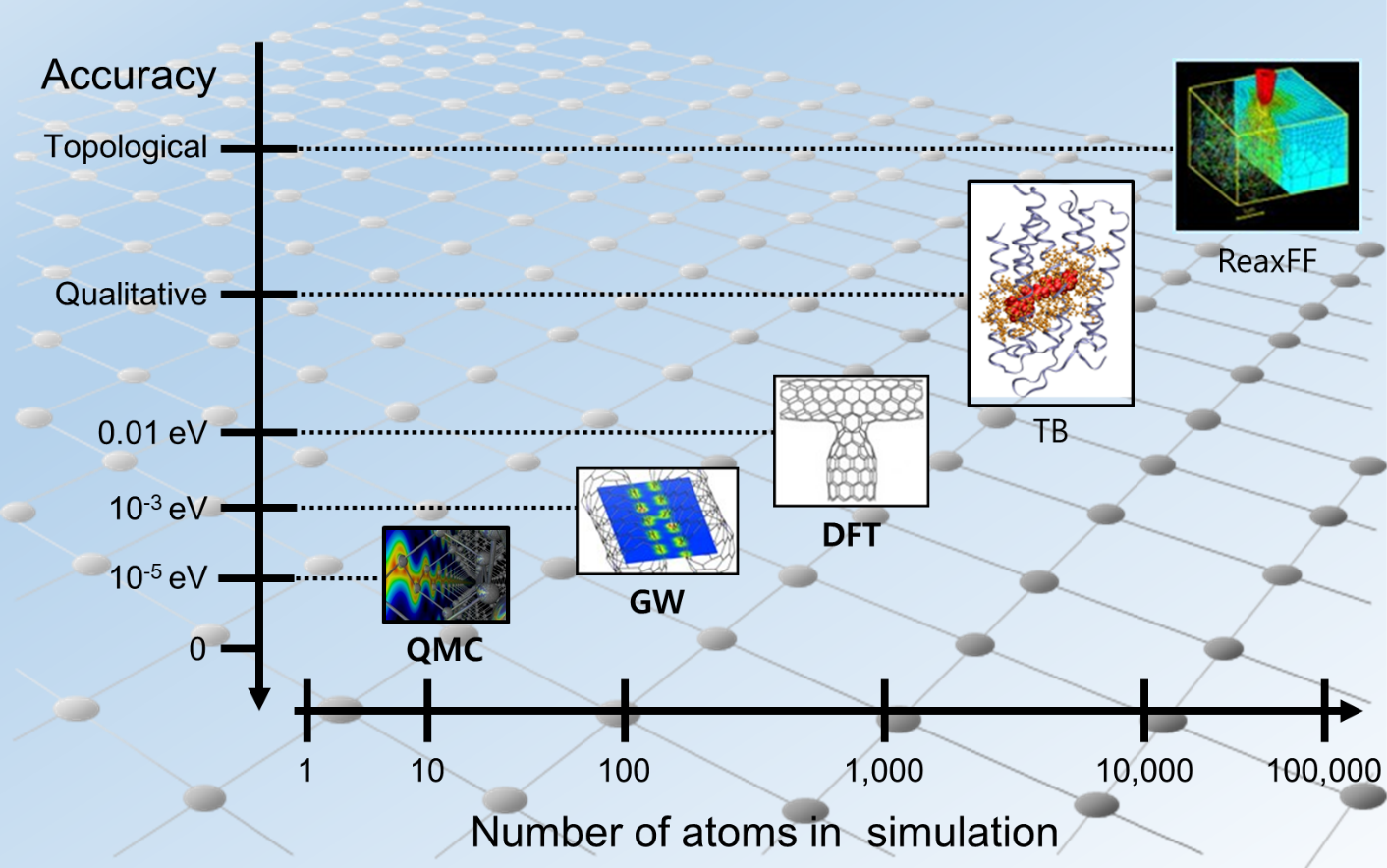
그림1. 각 시뮬레이션 방법이 다룰 수 있는 원자 개수와 정확도
| QMC | QMC method는 quantum monte carlo의 약자로 Schrödinger 방정식의 해를 구하는 방법의 하나이다. DFT나 GW 계산과 달리 특별한 가정이나 근사없이 many-body wavefunction을 직접 구할 수 있다. monte carlo는 도박으로 유명한 모나코의 도시 이름으로 그 이름에 걸맞게 QMC도 many-body wavefunction을 random하게 생성하여 Schrödinger 방정식의 해를 찾는다. 계산코드로는 CASINO등이 있다. |
|---|---|
| 장점 | 가장 정확한 계산 |
| 단점 | 가장 높은 수준의 계산자원 필요 |
| GW | GW method는 Schrödinger 방정식의 근사해를 구하는 방법의 하나로 DFT 계산의 단점인 exchange-correlation energy를 정확히 계산하기 위해 개발되었다. Green’s function formalism을 통해 Dyson’s equation을 풀어 더욱 엄밀한 계산을 수행한다. GW의 G는 Green’s function을 의미하고 W는 screened Coulomb potential을 의미한다. 계산코드로 BGW, GPAW, VASP 내장코드 등이 있다. |
| 장점 | band gap, absorption coefficient와 같은 광학적 성질계산에 활용 |
| 단점 | W계산을 위해 DFT보다 많은 계산자원을 필요 |
| DFT | DFT method는 density functional theory의 약자로 Schrödinger 방정식의 근사해를 구하기 위해 Khon-Sham equation을 푼다. 계산을 진행할 때 전자들의 파동함수대신 전자들의 밀도 함수로 대체하여 계산 함으로서 기존에 요구되었던 계산 자원보다 훨씬 적은 계산 자원을 통해 전자 수준의 시스템 계산이 가능하게 되었다. 계산코드로는 VASP, QE, CASTEP 등이 있으며 이를 통하여 전자 구조, 광학적 성질, 기계적 성질, 안정한 결정구조, 결함의 성질, 화학적 반응 등을 예측할 수 있다.장점: 전자수준의 계산을 비교적 낮은 계산자원으로 계산가능. |
| 단점 | self-interaction 문제가 있고 localized electron 기술 어려움 |
| EMTO-CPA | EMTO-CPA는 Schrödinger 방정식의 근사해를 구하는 방법의 하나로 Exact Muffin-Tin Orbital과 Coherent Potential Approximation의 약자이다. 기존의 KKR method를 기본으로 하며 Optimized Overlapping Muffin-Tin(OOMT)를 도입하여 kinetic energy의 정확성을 높였다. 또한 CPA근사로 High Entropy Alloy와 같이 조성이 복잡한 물질을 효율적으로 계산한다. 최근 단순한 CPA를 넘어 원소 간 local ordering을 포함하는 방향으로 발전하고 있다. 계산코드로는 Vitos 그룹의 FCD-EMTO-CPA등이 있다. | 장점 | band gap, absorption coefficient와 같은 광학적 성질계산에 활용 |
| 단점 | W계산을 위해 DFT보다 많은 계산자원을 필요 |
| AIMD | AIMD (ab-initio molecular dynamics)는 분자 동역학 계산에 있어 필요한 원자간의 force를 제일원리 계산을 통하여 생성하고 뉴턴 법칙에 따른 분자 동역학 계산을 진행하여 분자와 원자의 움직임, 온도에 따른 물성의 변화 등을 예측해 내는 것이다. 제일원리 계산을 통해 원자간의 force를 계산하기 때문에 다른 실험적인 값에 의한 potential이 필요 없다. 시간에 따른 원자의 움직임으로 인하여 전자의 분포가 변화하기 때문에 기존의 분자동역학으로 알 수 없는 물성을 예측가능하다. 하지만 제일원리계산을 이용하여 전자를 직접 기술하여 원자간의 force를 계산하기 때문에 많은 비용과 오랜 시간이 걸린다. |
|---|---|
| TB | TB는 tight binding의 약자로 LACO의 고체버전으로 생각할 수 있다. 원자 오비탈로부터 오비탈간 상호작용을 고려한 model Hamiltonian으로 전자 구조를 계산한다. 오비탈 간 상호작용이 직접적으로 드러나 있기 때문에 전자 구조 분석에 용이하다. 하지만 기본상수 외에 파라미터를 필요로 한다. 계산코드로는 DFTB+등이 있다. |
| 장점 | 낮은 계산자원 필요 |
| 단점 | hopping parameter 의존 |
| ReaxFF | ReaxFF는 reactive force field라고 부르며 제일원리계산과 다른 원자단위계산이다. 기존의 interatomic potential과 달리 bond order라는 개념을 도입하여 화학반응을 시뮬레이션하는데 사용된다. 주로 유기반응을 잘 시뮬레이션하며 전이금속이나 이온성 물질의 경우도 시뮬레이션 가능하지만 퍼텐셜생성에 어려움이 있다. |
| 장점 | 큰 스케일의 시뮬레이션이 가능 |
| 단점 | 다른 원자단위 시뮬레이션에 비해 많은 계산량을 요구 |
| NEB | NEB(nudged elastic band)는 반응물과 생성물사이의 energetic saddle point와 반응이 일어나는 경로를 찾는 방법이다. 이를 통하여 원자단위에서의 반응이 일어나는 경로와 반응을 하기 위한 energy barrier등을 예측 할 수 있다. |
| 장점 | band gap, absorption coefficient와 같은 광학적 성질계산에 활용 |
| 단점 | W계산을 위해 DFT보다 많은 계산자원을 필요 |
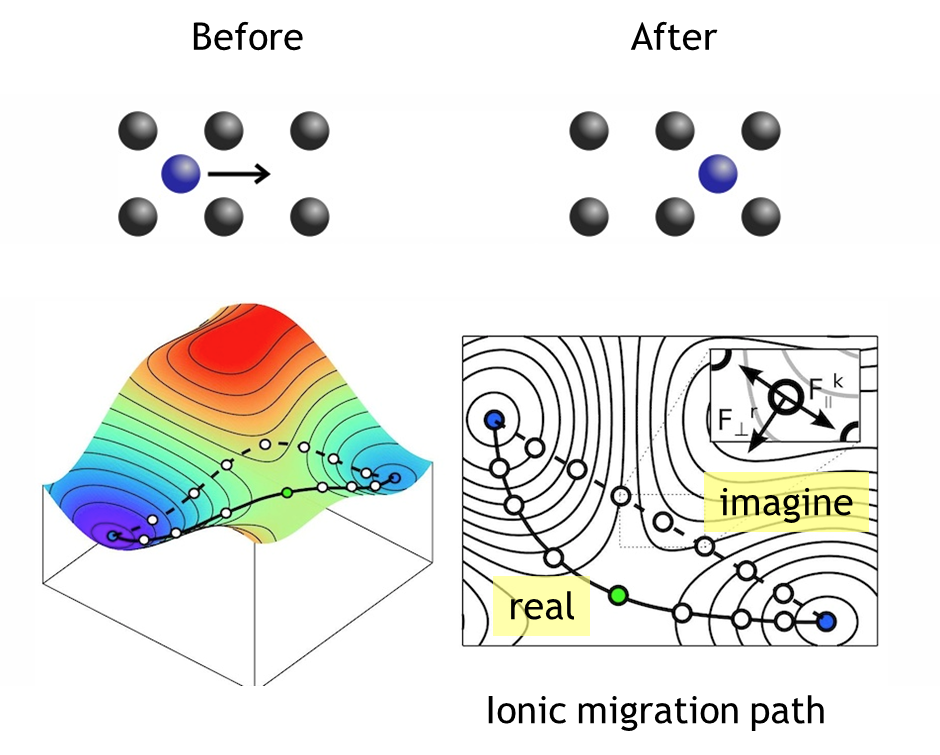
그림2. NEB를 통한 ionic migration path 예측
| USPEX | 진화 알고리즘과 제일원리, 분자 동역학 등을 이용하여 기존에 알려지지 않은 결정구조에 대하여 예측을 하는 code로 기존에 알려지지 않은 결정 구조와 상에 대한 예측이 가능하며 다른 압력에서 안정해지는 상 또한 계산을 통해 예측해 낼 수 있다. |
|---|
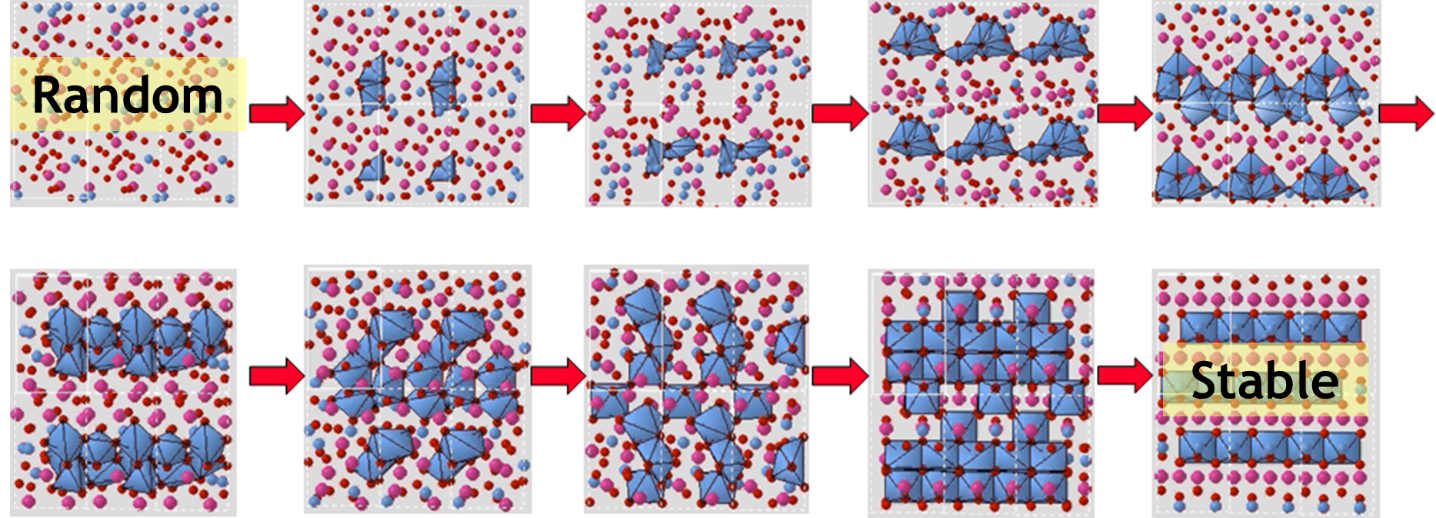
그림3. 진화 알고리즘을 이용하는 USPEX을 통한 새로운 결정구조 예측
